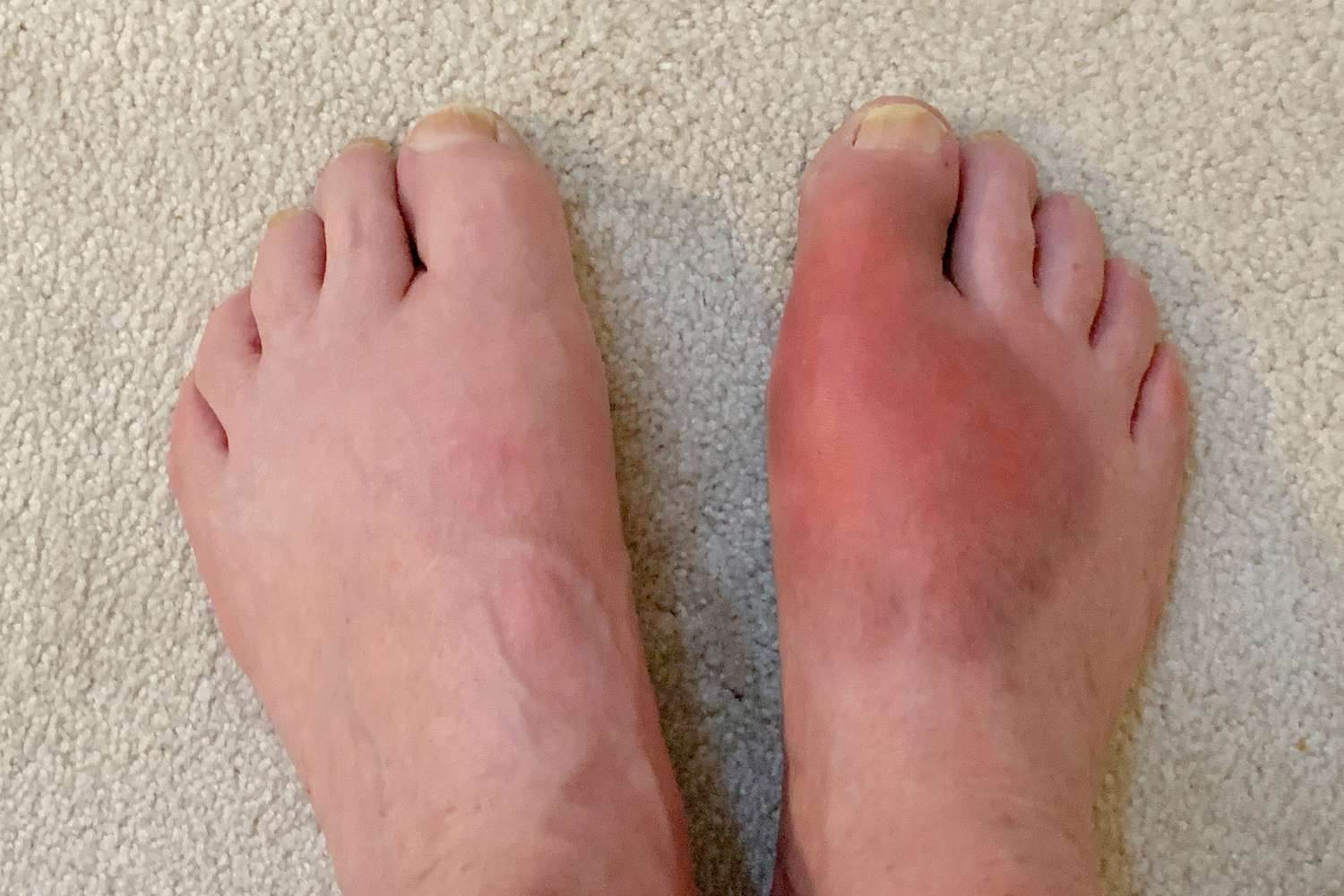
Gout is a condition often dismissed in popular culture with historical jokes, yet for those who suffer from it, the reality is a profoundly debilitating experience characterized by excruciating, sudden-onset joint pain. It is an inflammatory arthritis, but one fundamentally rooted in a metabolic disorder: chronic hyperuricemia, or an excess of uric acid in the blood. When uric acid levels become supersaturated, they precipitate, forming sharp, needle-like monosodium urate (MSU) crystals that deposit within the joints, synovium, and surrounding tissues. This deposition triggers an intense inflammatory response, typically attacking the joint at the base of the big toe (podagra) but capable of affecting any joint, and even causing chronic kidney issues. Effective management of gout requires a dual-pronged strategy that addresses both the immediate, agonizing inflammatory flare and the long-term, underlying hyperuricemia responsible for the disease’s persistence and progression. Understanding this distinction is the cornerstone of successful therapy, moving beyond temporary pain relief toward disease modification and crystal dissolution.
The reality is a profoundly debilitating experience characterized by excruciating, sudden-onset joint pain
The genesis of gout lies in the metabolic pathway of purines, compounds found naturally in the body and in various foods. When purines are metabolized, uric acid is produced as a waste product. Normally, the kidneys efficiently excrete this acid, maintaining a healthy balance. In individuals susceptible to gout, either the body overproduces uric acid (a small percentage of cases) or, far more commonly, the kidneys are unable to adequately excrete it (underexcretion). This sustained imbalance leads to hyperuricemia. The process of crystal deposition is subtle and silent, occurring years before the first acute flare. The immune system, recognizing the sharp MSU crystals as foreign invaders, launches an aggressive inflammatory assault involving specialized white blood cells (neutrophils). It is this explosive, misguided immune response, rather than the crystal deposition itself, that generates the hallmark pain, redness, swelling, and heat of an acute gout attack, turning a subtle metabolic problem into a crippling clinical event.
The genesis of gout lies in the metabolic pathway of purines, compounds found naturally in the body and in various foods
Acute gout flares demand rapid, decisive anti-inflammatory treatment to quell the agonizing pain and prevent structural damage to the joint. The choice of agent is dictated by the patient’s co-morbidities and the timing of intervention. Non-steroidal anti-inflammatory drugs (NSAIDs), such as indomethacin or naproxen, are highly effective if started at the very first sign of an attack. However, their use must be limited or avoided entirely in patients with peptic ulcer disease, heart failure, or significant kidney impairment. Colchicine, an older anti-inflammatory agent, is also highly effective but must be administered within the first 12 to 24 hours of symptom onset for maximal benefit and is often limited by gastrointestinal side effects. For patients where NSAIDs and colchicine are contraindicated or ineffective, corticosteroids—either orally administered (e.g., prednisone) or injected directly into the affected joint—provide the most potent anti-inflammatory relief. This phase of treatment is purely symptomatic; it addresses the fire, but does nothing to reduce the underlying fuel supply of uric acid.
Acute gout flares demand rapid, decisive anti-inflammatory treatment to quell the agonizing pain
The critical shift in managing gout is the pivot from treating the flares to managing the disease itself through long-term urate-lowering therapy (ULT). The primary goal of ULT is to achieve and maintain a sustained serum uric acid (sUA) level below the saturation point for MSU crystals, typically defined as $6.0\text{ mg/dL}$. In patients with severe, chronic manifestations like tophi (large crystalline deposits in soft tissues), the target is even lower, often $5.0\text{ mg/dL}$. This aggressive targeting is necessary because only by maintaining these low sUA levels can the body dissolve the existing crystal deposits that have accumulated over years. Allopurinol, a xanthine oxidase inhibitor (XOI), remains the most commonly prescribed and foundational ULT. It works by blocking the enzyme responsible for converting purines into uric acid, thereby reducing the total production of the acid.
The primary goal of ULT is to achieve and maintain a sustained serum uric acid level below the saturation point for MSU crystals
Initiating allopurinol requires a careful titration process. Unlike acute medications, allopurinol is typically started at a low dose and gradually increased over weeks or months until the target sUA level is consistently achieved. This methodical approach is critical, as rapidly lowering uric acid levels can paradoxically precipitate an acute gout flare, due to the mobilization of existing crystals. Furthermore, a rare but severe hypersensitivity syndrome is associated with allopurinol, particularly in patients with kidney impairment and certain genetic markers, necessitating caution and monitoring. For patients who fail to achieve target sUA levels on maximum doses of allopurinol, or who experience intolerable side effects, Febuxostat (another XOI) offers an alternative pathway, though its use requires careful consideration due to potential cardiovascular risks identified in certain studies.
Initiating allopurinol requires a careful titration process
In a smaller subset of patients, underexcretion remains the dominant problem even after maximal XOI therapy. For these individuals, uricosuric agents—such as probenecid—may be added or utilized. These drugs work by increasing the kidneys’ ability to excrete uric acid into the urine. Probenecid is primarily effective in patients who have a robust kidney function remaining, as its mechanism relies on renal clearance. However, these agents increase the risk of kidney stones, demanding meticulous patient hydration and careful monitoring of urine output. For the most severe, refractory cases of gout—those with large, debilitating tophi or severe joint damage who have failed oral therapies—biologic agents like pegloticase, an intravenous enzyme that rapidly breaks down uric acid, are reserved. Pegloticase is a highly effective, but high-risk therapy, often requiring pre-treatment with anti-inflammatory agents to prevent severe infusion reactions and flare-ups associated with rapid crystal dissolution.
For these individuals, uricosuric agents—such as probenecid—may be added or utilized
A crucial and often misunderstood facet of ULT is the need for concurrent anti-inflammatory prophylaxis when starting urate-lowering drugs. Paradoxically, the very act of lowering uric acid often triggers a gout attack as crystals begin to dissolve and mobilize from the joint space. Therefore, patients starting allopurinol or Febuxostat are typically prescribed a low dose of colchicine or a daily NSAID for the first three to six months. This prophylactic regimen acts as a protective shield, dampening the immune response during the period of crystal mobilization, ensuring patient comfort and compliance with the necessary, long-term urate-lowering goal. Cessation of this prophylaxis should only occur after the target sUA level has been maintained for a sustained period, indicating the stability of the dissolved crystal environment.
The very act of lowering uric acid often triggers a gout attack as crystals begin to dissolve and mobilize from the joint space
While pharmacotherapy is the bedrock of gout management, lifestyle and dietary modifications play a significant, though secondary, role. For decades, gout management was dominated by strict dietary restrictions, particularly avoiding organ meats, specific seafood, and excessive alcohol consumption, all of which are high in purines. While these restrictions can help, they are usually insufficient to control hyperuricemia alone, which is primarily driven by renal excretion issues. Modern recommendations emphasize reducing intake of high-fructose corn syrup, sugar-sweetened beverages, and alcohol (especially beer and spirits), as these directly impair renal uric acid excretion. Furthermore, weight loss—especially a gradual, sustained loss—can improve uric acid clearance and reduce systemic inflammation. Hydration is also paramount, as adequate fluid intake aids in flushing uric acid through the kidneys, reducing the risk of crystallization in the urinary tract.
Lifestyle and dietary modifications play a significant, though secondary, role
Gout is a disease with a significant cardiovascular and renal component that extends far beyond joint inflammation. Chronic hyperuricemia and chronic gout are often associated with hypertension, chronic kidney disease (CKD), and increased risk of cardiovascular events. This systemic link necessitates a comprehensive approach to managing all co-morbidities. The primary care physician and the rheumatologist must work in concert to ensure that hypertension is aggressively treated, that kidney function is preserved, and that lipid profiles are managed. Failure to address these associated conditions means that even if the gout is controlled, the patient’s overall health and prognosis remain compromised. Viewing gout as an isolated musculoskeletal issue is a critical clinical error that neglects its true systemic nature.
Gout is a disease with a significant cardiovascular and renal component that extends far beyond joint inflammation
The transition from flare management to disease management requires a significant psychological shift for the patient. Many patients, once the acute agony of a flare subsides, become lax in their adherence to daily ULT, mistakenly believing the disease is cured. Continuous patient education is essential, reinforcing that gout is a chronic, progressive disease requiring indefinite treatment to keep crystal deposits dissolved and sUA levels low. The success of therapy is not measured by the absence of flares today, but by the continuous, low sUA level that prevents crystal deposition over the next five to ten years, protecting both joints and major organs. Regular sUA monitoring (every few months until the target is met, then less frequently) serves as the objective, non-symptomatic measure of treatment success, providing tangible proof of the drug’s efficacy and justifying the commitment to daily medication.